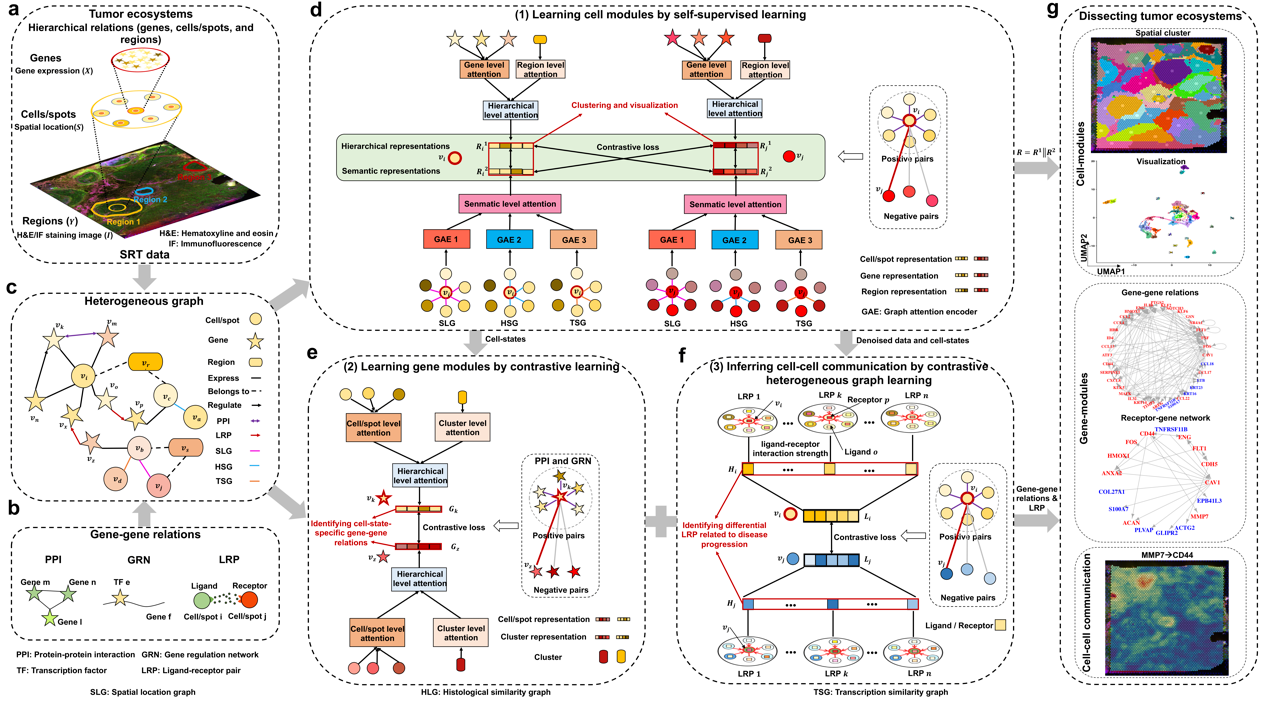
Overview of the stKeep model [IMAGE: SIBCB]
Cancer is a tumor ecosystem where cancer cells interact with surrounding immune and stromal cells to survive in harsh conditions. Cell states are influenced by both intracellular molecular networks and intercellular CCCs. Spatial omics technologies provide transcriptomics profiles with spatial locations and pathological images, offering new opportunities to dissect the tumor microenvironment (TME) by analyzing the molecular mechanisms driving disease. However, the lack of computational tools for exploring the complex relations between cells, genes, and histological regions has hindered the interpretation of the complex structure of the TME.
In a study published in Nature Communications, the teams led by Professor Chen Luonan from the Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology (SIBCB) of the Chinese Academy of Sciences, collaborated with Dr. Zuo Chunman from Donghua University to propose a novel graph method called stKeep that elucidates the heterogeneous TME by identifying cell-modules, gene-modules, and cell-cell communication (CCC) models.
In this study, the researchers introduced stKeep, a graph embedding method that integrates multimodal data (including histology, gene expression, spatial location, and histological regions) and molecular networks (such as gene regulatory networks, protein-protein interaction, and ligand-receptor pairs), to elucidate TME heterogeneity. Specifically, stKeep constructs a heterogeneous graph with three types of nodes (cells/spots, genes, and tumor regions) and eight links to characterize TME.
It then (1) utilizes multi-layer relational graph embedding and contrastive learning algorithms to aggregate information from related genes, tumor regions, and semantically connected cells, computing cell-modules to detect more refined cell-states; (2) integrates information from relevant cells, cell-states, and gene-gene relations to compute gene-models, identifying cell-state-specific gene-gene interactions; and (3) quantifies single-cell/spot communication strength using an attention mechanism, inferring CCC patterns that reflect cell-state differences within the TME and identifying ligand-receptor pairs associated with disease progression through contrastive learning techniques.
In analysis of triple-negative breast cancer, stKeep detects more cell states within tumor regions than other methods. By integrating paired scRNA-seq data, stKeep identifies a group of myoepithelial cells, previously misclassified as normal cells in earlier studies, as tumor cells. It also infers key transcription factors, ligands, and receptors in these cells that contribute to disease progression.
In analysis of colorectal cancer liver metastasis samples, researchers also identified key cell populations and CCC mechanisms involved in the metastasis of colon cancer cells to normal liver tissue. These findings were further validated through independent samples and clinical data.
This study proposed a novel method, stKeep, which integrates multimodal and molecular network data to analyze tumor ecosystems, facilitating new applications of spatial transcriptomics data in clinical prognosis and immunotherapy.
For more information, please contact:
Professor Chen Luonan
E-mail: lnchen@sibcb.ac.cn
Center for Excellence in Molecular Cell Science,
Shanghai Institute of Biochemistry and Cell Biology,
Chinese Academy of Sciences
Source: Center for Excellence in Molecular Cell Science,
Shanghai Institute of Biochemistry and Cell Biology,
Chinese Academy of Sciences

