CHINESE ACADEMY OF SCIENCES

-

- SARS-CoV-2 evolution and human transmission mechanism revealed
- (NO.160 February 2020)
- Issue NO:Research Progress Updated:30-04-2020
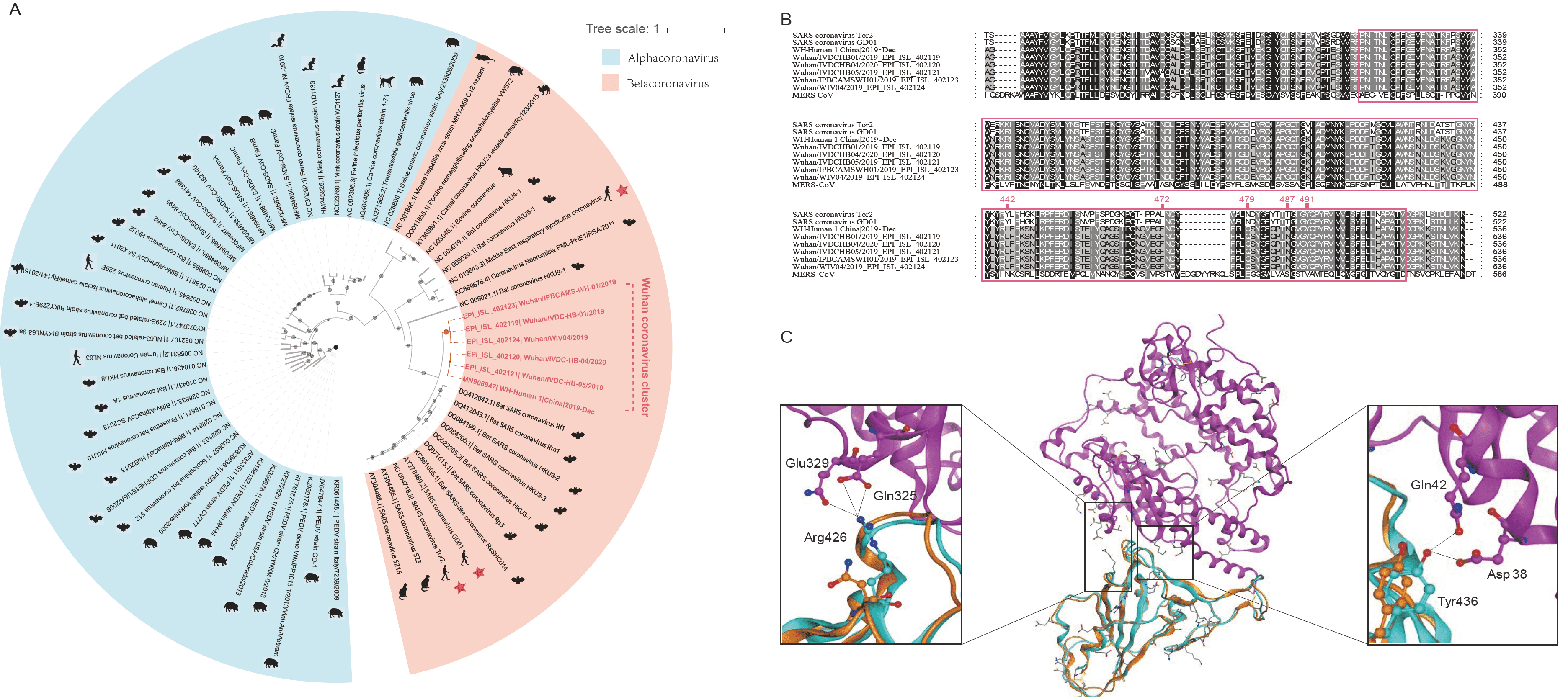
Evolutionary analysis of the coronaviruses and modeling of the SARS-CoV-2 S-protein interacting with human ACE2.
(A) Phylogenetic tree of coronaviruses based on full-length genome sequences;
(B) Amino acid sequence alignment of the RBD domain of coronavirus S-protein;
(C) Structural modeling of the SARS-CoV-2 (WH-human_1 as representative) S-protein complexed with human ACE2 molecule.----
A research work entitled “Evolution of the Novel Coronavirus from the Ongoing Wuhan Outbreak and Modeling of Its Spike Protein for Risk of Human Transmission” was published online in SCIENCE CHINA Life Sciences on January 21, 2020. Doctor Hao Pei (Institut Pasteur of Shanghai, CAS), Doctor Zhong Wu (Beijing Institute of Pharmacology and Toxicology), and Doctor Li Xuan (CAS Center for Excellence in Molecular Plant Sciences, CAS) are co-correspondence authors of this paper. Xu Xintian, Chen Ping, and Wang Jingfang are the co-first authors.
The occurrence of concentrated pneumonia cases in Wuhan City, Hubei Province, China was first reported on December 30, 2019 by the Wuhan Municipal Health Commission. The pneumonia cases were found to be linked to a large seafood and animal market in Wuhan. The Center for Disease Control and Prevention (CDC) and Chinese health authorities later determined and announced that a novel coronavirus, denoted as SARS-CoV-2, had caused the pneumonia outbreak. The current public health emergency partially resembles the emergence of the SARS outbreak in Southern China in 2002. Both happened in winter, with initial cases linked to exposure to live animals sold at animal markets, and both were caused by previously unknown coronaviruses.
The first genome sequence of the SARS-CoV-2 was released on January 10, 2020, and subsequently multiple additional SARS-CoV-2 genome sequences were released. To understand the origin of the SARS-CoV-2 and its genetic relationship with other coronaviruses, the authors performed phylogenetic analysis on the collection of coronavirus sequences from various sources. The results showed the SARS-CoV-2 were clustered together in the phylogenetic tree, which belong to the Betacoronavirus genera (Fig A). It is likely that their natural hosts are bats. They and the SARS/SARS-like coronaviruses shared a common ancestor that resembles the bat coronavirus HKU9-1.
To investigate the SARS-CoV-2 and its interaction with its hosts, the authors looked into the RBD domain of the spike protein (S-protein) of the SARS-CoV-2, which had several patches of sequences with a high homology to that of SARS-CoV_Tor2 and HP03-GZ01 (Fig B). The residues at positions 442, 472, 479, 487 and 491 in SARS-CoV S-protein were reported to be at receptor complex interface and considered critical for cross-species and human-to-human transmission of SARS-CoV. However, four of the five critical residues are not preserved except Tyr491 in S-protein of the SARS-CoV-2.
To assess the risk of human transmission of the SARS-CoV-2, the authors performed structural modeling of its S-protein and evaluated its ability to interact with human ACE2 molecules. The computational model of the SARS-CoV-2 S-protein showed a Cα RMSD of 1.45 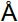 on the RBD domain compared to the SARS-CoV S-protein structure (Fig C). The binding free energy between the SARS-CoV-2 S-protein and human ACE2 was -50.6 kcal/mol, whereas that between SARS-CoV S-protein and ACE2 was -78.6 kcal/mol. Our result points to the important discovery that the RBD domain of the SARS-CoV-2 S-protein supports strong interaction with human ACE2 molecules despite its sequence diversity with SARS-CoV S-protein. Thus the SARS-CoV-2 poses a significant public health risk for human transmission via the S-protein - ACE2 binding pathway.
on the RBD domain compared to the SARS-CoV S-protein structure (Fig C). The binding free energy between the SARS-CoV-2 S-protein and human ACE2 was -50.6 kcal/mol, whereas that between SARS-CoV S-protein and ACE2 was -78.6 kcal/mol. Our result points to the important discovery that the RBD domain of the SARS-CoV-2 S-protein supports strong interaction with human ACE2 molecules despite its sequence diversity with SARS-CoV S-protein. Thus the SARS-CoV-2 poses a significant public health risk for human transmission via the S-protein - ACE2 binding pathway.
The article “Evolution of the Novel Coronavirus from the Ongoing Wuhan Outbreak and Modeling of Its Spike Protein for Risk of Human Transmission” gave evidence of the possibility of human transmission of the current SARS-CoV-2 and subsequent public health risk. The authors hope these joint results will contribute to the common good of all people and all parties involved.
This work was supported in part by grants from the National Science and Technology Major Projects for “Major New Drugs Innovation and Development” (directed by Doctor Li Song) (2018ZX09711003) of China, the National Key R&D Program (2018YFC0310600) of China, the National Natural Science Foundation of China (31771412), and Special Fund for strategic bio-resources from Chinese Academy of Sciences (ZSYS-014). The authors also acknowledge the National Institute for Viral Disease Control and Prevention, China CDC; Wuhan Institute of Virology, Chinese Academy of Sciences; Institute of Pathogen Biology, Chinese Academy of Medical Sciences & Peking Union Medical College; and Wuhan Jinyintan Hospital for their efforts in research and collecting the data and genome sequencing sharing.
For more information, please contact:
Professor Hao Pei
Institut of Pasteur of Shanghai,Chinese Academy of Sciences
E-mail: phao@ips.ac.cn
Source: Institut Pasteur of Shanghai, Chinese Academy of Sciences