CHINESE ACADEMY OF SCIENCES

-

- Better coverage database for human genome variation developed
- (NO.158 December 2019)
- Issue NO:Research Progress Updated:30-04-2020
A recent study led by Dr. Xu Shuhua from the CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health of the Chinese Academy of Sciences (CAS), created a genome variation resource/database, PGG.SNV (https://www.pggsnv.org) which archives 265 million single nucleotide variations (SNVs) across 220,147 present-day genomes and 1,018 ancient genomes, including 1,009 newly sequenced genomes, representing 977 global populations.
PGG.SNV significantly improves the coverage of Asian populations which are significantly under-represented in other available database such as gnomAD. Compared with the available database, another unique feature of the PGG.SNV is that it provides an estimation of population genetic diversity and evolutionary parameters.
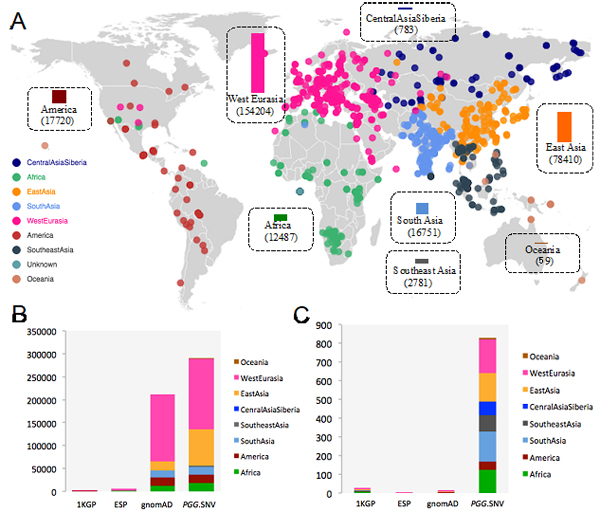
Figure 1: An overview of geographical distribution of human populations covered by PGG.SNV genome data (A)The distribution of human populations covered by the PGG.SNV database and the statistical number of genomes; (B) Comparison of PGG.SNV and gnomAD etc. in terms of genome numbers; (C) Comparison of PGG.SNV and gnomAD etc. in terms of number of populations [Image: Dr. Xu Shuhua’s group]
Despite Asia being the Earth’s largest and most populous continent, most of the genomic studies have been conducted in Europe and the United States. Accordingly, currently available human genome variation resources are based on populations of European ancestry. For example, nearly half of the genomes in gnomAD are of European ancestry and merely 9% of the genomes are of African ancestry, resulting in an enormous number of variants harbored in Asian genomes that cannot be observed in the extensively studied populations of European ancestry. Moreover, samples in gnomAD were merely classified into groups, mainly on the continental level, leaving the majority of the specific ethnic groups unknown. For example, gnomAD groups East Asians roughly into three categories: “Korean”, “Japanese” and “other East Asians”. Therefore, researchers fail to query the allele frequencies for most East Asian populations.
Compared to other frequently used data sets, PGG.SNV documents more genomes and represents a much more comprehensive genomic diversity of worldwide populations. For instance, there are 90,514 Asian genomes included in PGG.SNV, compared to 993 and 25,285 in the 1KGP and gnomAD data sets, respectively. Moreover, PGG.SNV includes 1,009 newly-generated whole genome sequences from 16 ethnic groups, especially many indigenous groups living in East Asia and Southeast Asia whose genomes have not been sequenced before. Beside present-day human populations, the database integrates 1,018 ancient genomes that represent time periods from the 430,000 years before the present day up to the early 20th century, which is rarely considered in many other existing databases.
With a comprehensive catalogue of genetic variants and annotations, PGG.SNV enables studies of variants that are rare or not existing in well-studied populations, provides the population prevalence of variants in various populations with little ancestral bias and further guides Mendelian-inherited disease mapping studies. PGG.SNV documents many ancient genomes and compares them with contemporary human genomes, allowing researchers to understand the evolutionary trajectory of genetic variants as well as gene flow or introgression events. Moreover, this database improves interpretations of putative causal loci for Mendelian diseases, population differentiation analysis, and understanding of adaptation to local environments for global populations. Eventually, PGG.SNV will help advance our understanding of the biological meaning of the human genome sequence in light of human evolution.
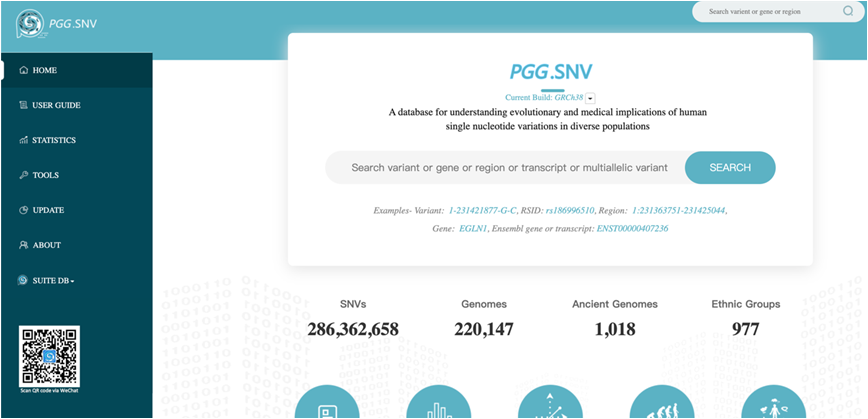
Figure 2: The main user interface of the PGG.SNV [Image: Dr. Xu Shuhua’s group]
PGG.SNV provides a web-based user interface to access data. The users can search genetic variants by physical position, RSID, a genomic region, official gene symbol or Ensembl gene name etc. PGG.SNV has also embedded a web-based tool (https://www.pggsnv.org/tools.html) for the generation of figures after users have uploaded their own analyses. In addition to the web-based interface, users can query variants using a mobile application (App) by linking to the WeChat official account named PGGbase.
The study was published online in Genome Biology on October 22, 2019, entitled “PGG.SNV: Understanding the evolutionary and medical implications of human single nucleotide variations in diverse populations”.
This work was conducted by Dr. Zhang Chao, Dr. Lu Yan, and PhD students Gao Yang from ShanghaiTech University, Ning Zhinlin and a few members from Dr. Xu Shuhua’s team.
For more information, please contact:
Wang Jin (Ms.)
Shanghai Institute of Nutrition and Health,
Chinese Academy of Sciences
E-mail: sibssc@sibs.ac.cn
Source: Shanghai Institute of Nutrition and Health,
Chinese Academy of Sciences