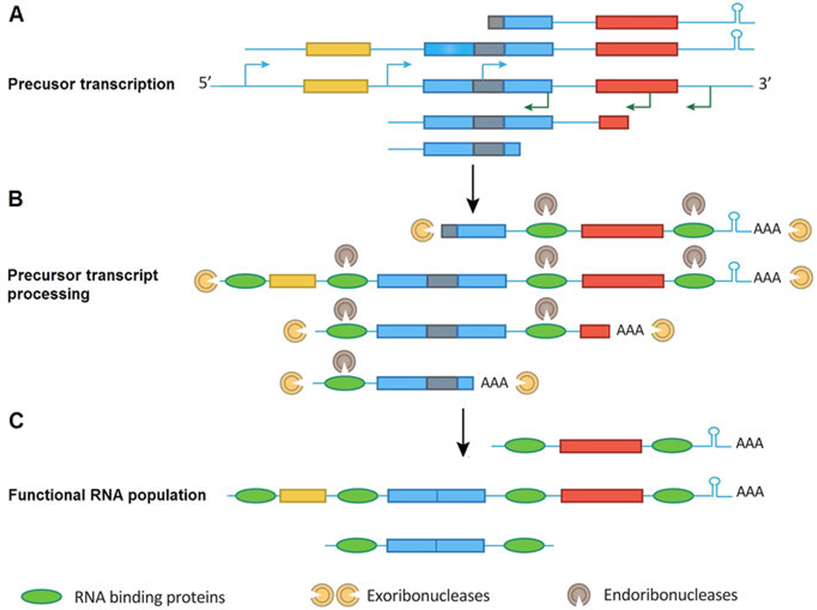
Genome-wide transcriptions of the eukaryotes are incredibly complex. Widespread bidirectional promoters generate pervasive genome transcription, and transcriptions can originate from both genic and intergenic regions that have no well-defined functional elements, resulting in substantial transcription of long (>200 bp) and short (<200 bp) RNAs. The long precursor RNAs (both coding and noncoding) can be further processed into shorter RNAs. Together, these processes generate an unexpected genome transcriptional output. This eukaryote transcription complexity has been well studied in yeast, Drosophila, and human cells, but it remains poorly understood in prokaryotes, such as plastids, leading to the idea that only eukaryotes harbor complex genome transcription and procession systems.
Despite living in host eukaryotic cells for approximately 1 billion years since the endosymbiosis event, the plastid still preserves its prokaryotic characteristics. Previous studies suggested some prokaryotic features of plastids (e.g., prokaryotic-type gene promoters and terminators and clustered gene transcripts). It has long been considered that some chloroplast (cp) functional genes are transcribed as polycistronic transcripts that are subsequently processed into small mature RNAs, potentially indicating limited transcriptional units within the plastome (about 20 major transcriptional units) and many of these un-transcribed regions (e.g., regions between two transcription units; ≥40percentpercent of all genomic regions). Under such a polycistronic operon transcription model, plastome genes would be transcribed from intrinsic promoters and later form stable, size-fixed transcripts. However, this model cannot account for all the transcriptional products at whole-genome level, such as tremendous plastid noncoding RNA output, pseudogene transcription, multiple alternative promoters/terminators, numerous heterogeneous and overlapping transcript isoforms, and gene transcription uncoupling in the same polycistron. These transcriptional dynamics and heterogeneity suggest that an additional general transcriptional mechanism triggers whole plastome transcription.
Prokaryotes possess a simple genome transcription system that is different from that of eukaryotes. In chloroplasts (plastids), it is believed that the prokaryotic gene transcription features govern genome transcription. However, the polycistronic operon transcription model cannot account for all the chloroplast genome (plastome) transcription products at whole-genome level, especially regarding various RNA isoforms. By systematically analyzing transcriptomes of plastids of algae and higher plants, and cyanobacteria, we find that the entire plastome is transcribed in photosynthetic green plants, and that this pattern originated from prokaryotic cyanobacteria — ancestor of the chloroplast genomes that diverged about 1 billion years ago. The research group headed by Prof. Gao Lizhi of the Germplasm Bank of Wild Species of the Kunming Institute of Botany, Chinese Academy of Sciences(CAS), proposes a multiple arrangement transcription model that multiple transcription initiations and terminations combine haphazardly to accomplish the genome transcription followed by subsequent RNA processing events, which would explain the full chloroplast genome transcription phenomenon and numerous functional and/or aberrant pre-RNAs. Their findings indicate a complex prokaryotic genome regulation when processing primary transcripts.
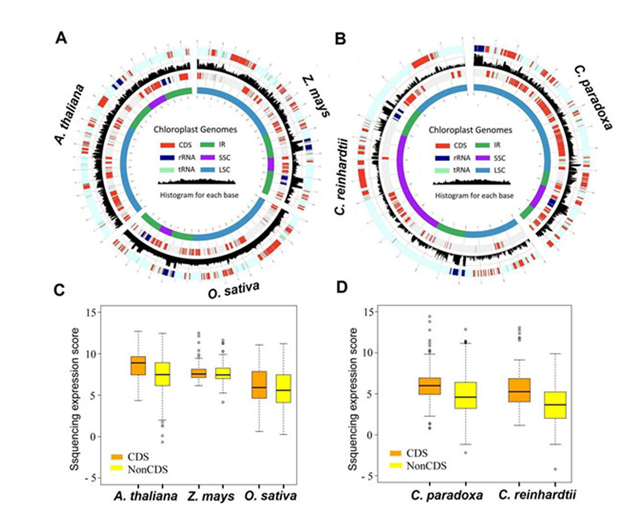
In plastids and bacteria, polyadenylation of the precursor transcripts serves as a necessary process for precise cleavage of functional RNAs and rapid degradation of non-functional RNAs. Thus, the assessment of polyA+ transcripts is suitable for the analyses of RNA metabolisms in plastids because it takes account of mRNA processing and transcription. The total plant cell transcriptome includes both nuclear and organelle (chloroplast and mitochondrion) transcripts, while traditional transcriptome analyses only focus on nuclear transcripts. They first isolated the plastid transcriptome (p-transcriptome) data from the total transcriptomes for three higher plants, rice (Oryza sativa), maize (Zea mays), Arabidopsis(Arabidopsis thaliana), one green algae Chlamydomonas (Chlamydomonasreinhardtii), and one basally diverging unicellular glaucophytes Cyanophoraparadoxa with recently published polyA+ transcriptome datasets. For the three higher plants, the transcriptome reads were from single tissue samples of shoots or leaves, except for Arabidopsis, which was from seedlings and flowers. The Chlamydomonas and glaucophytes reads were from cells cultured under normal conditions. After strict sequence quality control (See Experimental Procedures), transcriptome reads from each species, varying from 119 to 587 million, were further mapped to their own plastomes using a stringent pipeline.
Interestingly, the researchers found that the complete plastomes were covered by transcriptome reads (>99 percent for each species) with considerable read depths (from 480 to 47,875, depending on the total data). The transcriptome sequence reads may represent processed primary transcripts that are produced from precursor transcripts, with nearly full coverage of cptranscriptome reads mapped to the plastome, indicating the basal transcription nature of the entire plastomes of plants and algae. In Chlamydomonas, the initial genome coverage (about 91 percent) was relatively low. The Chlamydomonas plastome contains more than 20 percent repetitive sequences, and this may result in reduced coverage (only one location was allowed for reads mapping; see Experimental Procedures). Indeed, after removing the repeat sequences of the Chlamydomonas plastome, the coverage exceeded 99 percent. For all the examined species, intergenic regions were also hit by substantial sequence reads, only slightly lower than that for coding regions, further suggesting that the intergenic regions are highly transcribed and that the removal of intergenic regions is not necessary for the polyadenylation/degradation of plastid primary transcripts. Reads mapping resulted in a few unmapped regions (~1 percent of the total genome), of which >90 percent had a sequence length <30 bp. The researchers then validated the entire plastome transcription in rice by using reverse transcription polymerase chain reaction (RT-PCR) to confirm that all genomic regions they examined were indeed transcribed. Collectively, their transcriptome analyses provide direct evidence for whole-genome transcription in both green plants and algae.
The above achievement was published in Scientific Reports 6, Article number: doi: 10.1038/srep30135 (2016), tiled Full transcription of the chloroplast genome in photosynthetic eukaryotes. PhD Students Shi, C. and Xia Enhua are the first authors; Prof. Gao Lizhi is the corresponding author; collaborators include Wang Shuo from the Kunming University of Science and Technology, et al.

