CHINESE ACADEMY OF SCIENCES

-

- Cobalt-catalyzed sequential site-and stereoselective hydrosilylation of 1,3- and 1,4-enynes
- (NO.187 May 2022)
- Issue NO:Research Progress Updated:31-05-2022
Cobalt-catalyzed sequential site- and stereoselective hydrosilylation of 1,3- and 1,4-enynes [IMAGE: MENG FANKE]
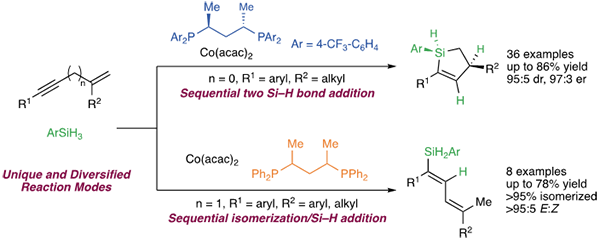
Alkenylsilanes are important building blocks in organic synthesis, as they can transform into other functional groups with versality. In addition, alkenylsilanes are of high stability and low toxicity. One of the most straightforward and atom-economic approaches for preparation of alkenylsilanes is metal-catalyzed hydrosilylation of alkynes. One of the challenges is to identify catalysts for accurate control of the number of the Si-H bonds involved in the reactions when multiple Si-H bonds exist in the silanes. Moreover, enantioenriched organosilanes bearing a Si-stereogenic center have attracted increasing attention in medicinal chemistry and material science, as the Si-stereogenic center can induce significant alteration of the biological and physicochemical properties. They are also important intermediates for stereoselective transformations. However, the cascade process of sequential site selective hydrosilylation of 1,3-enynes followed by diastereo- and enantioselective intramolecular hydrosilylation of the resulting dienylsilanes with simultaneous establishment of a carbon-stereogenic center and a silicon-stereogenic center remained undeveloped.
The Meng Fanke group of the State Key Laboratory of Organometallic Chemistry of the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, has been focusing on the study of novel cobalt-catalyzed asymmetric reactions. In previous research, they found that conjugated enynes bearing a 1,1-disubstituted alkene could undergo double hydrosilylation promoted by Xantphos-Co complex to afford cyclic silanes.
Recently, they have made new research progress. Unique reaction modes of Co-catalyzed hydrosilylation of enynes through Ojima-Crabtree isomerization of dienyl cobalt species were revealed. Under the circumstances of a simple chiral catalyst, sequential double hydrosilylation of enynes and primary silanes occurred to conform a C-stereogenic center and a Si-stereogenic center simultaneously with high chemo- and stereoselectivity.
It was found by the researchers that for 1,3-enynes and primary silanes, substrates bearing electron-withdrawing and -donating groups were tolerated, producing Si-stereogenic cyclic silanes with high efficiency and selectivity. The Si-H bonds of the products could undergo stereoselective transformations to generate tetrasubstituted silanes without racemization of the Si-stereogenic centers. When 1,4-unconjugated enynes were adopted, reaction occurred to provide dienyl silanes. Mechanistic studies showed that the substituent on the alkene moiety of the 1,3-enyne did facilitate the Ojima-Crabtree isomerization of (E)-dienyl cobalt species to reduce the 1,3-allylic strain. 1,4-Enyne would first undergo isomerization to generate a 1,3-conjugated enyne, followed by hydrosilylation of the alkyne.
The research outcome has been reported in JACS (DOI: 10.1021/jacs.2c00288).
For more information, please contact:
Ph.D. Professor Meng Fanke
E-mail: mengf@sioc.ac.cn
Shanghai Institute of Organic Chemistry,
Chinese Academy of Sciences
Source: Shanghai Institute of Organic Chemistry,
Chinese Academy of Sciences